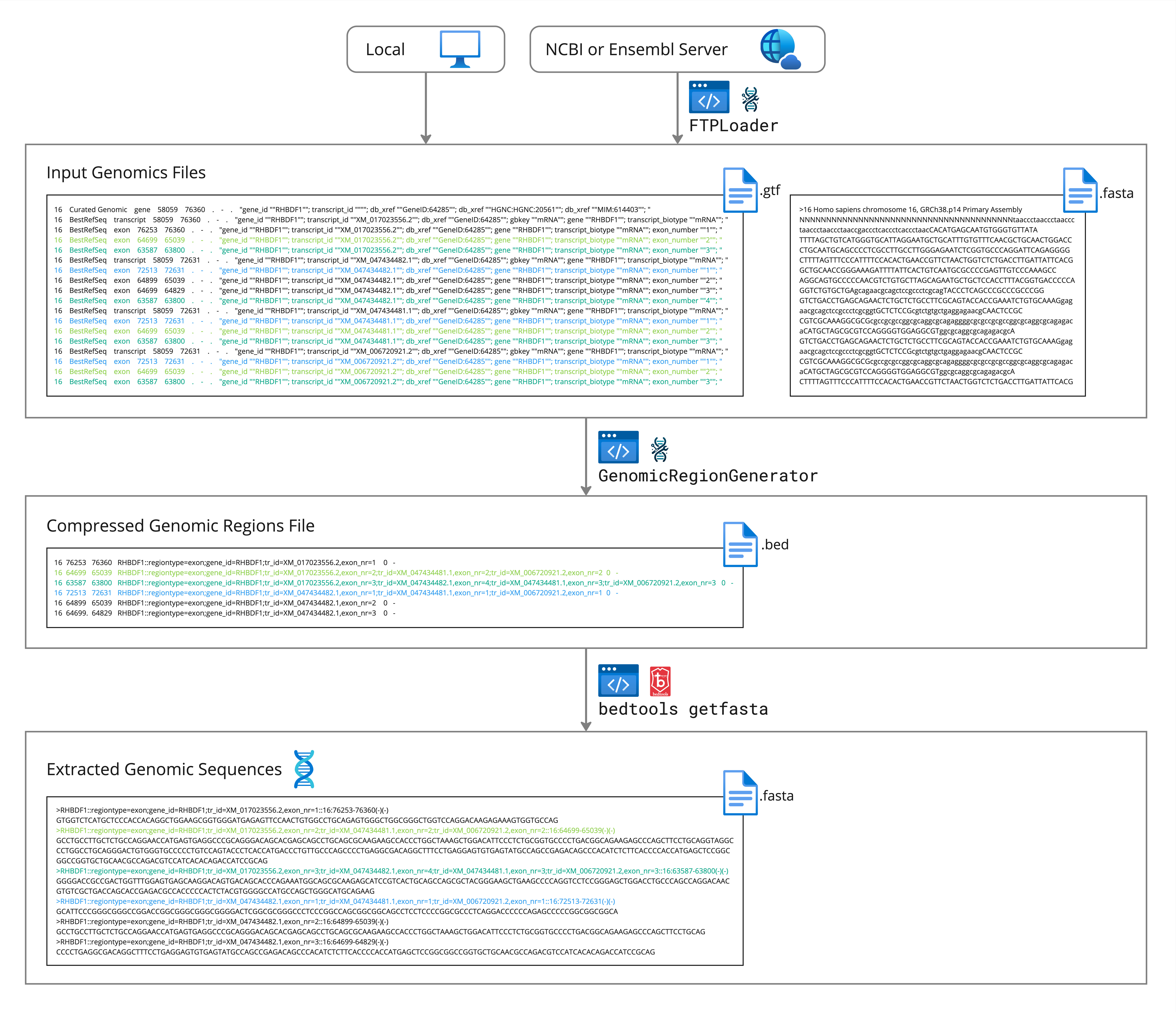
Genomic Region Generator
This pipeline is designed to extract genomic sequences of a specific type (e.g. exons, UTRs, etc.) from Fasta and GTF files. The required Fasta and GTF files can be automatically retrieved from NCBI or Ensemble, or provided as custom files by the user. If a custom reference is chosen, a GTF file with gene annotations and a Fasta file with the genome sequence have to be provided. When choosing a NCBI or Ensembl reference, the annotation GTF file and genome sequence Fasta file will be downloaded automatically via FTP from the respective servers. Therefore, the user has to define the species, annotation release and taxon (only for NCBI). From the given annotations, user-defined genomic regions are extracted. The genomic regions are stored in a compressed memory efficient format, which eliminates duplicated sequences stemming from common exons of different gene isoforms, while preserving the isoform information. The user can choose from a pre-defined list of genomic regions, i.e. intergenic, gene, CDS, exon, intron, 3’ UTR, 5’ UTR and exon-exon junctions.
Command-Line Call
To create sequences of genomic regions from NCBI annotations you can run the pipeline with
genomic_region_generator -c data/configs/genomic_region_generator_ncbi.yaml
where:
-c: config file, which contains parameter settings, specific to NCBI genomic region generation, genomic_region_generator_ncbi.yaml contains default parameter settings
All steps and config parameters will be documented in a log file, that is saved in the directory where the pipeline is executed from.
The logging file will have the format: log_genomic_region_generator_{year}-{month}-{day}-{hour}-{minute}.txt.